Machine Learning Classification of Local Environments in Molecular Crystals
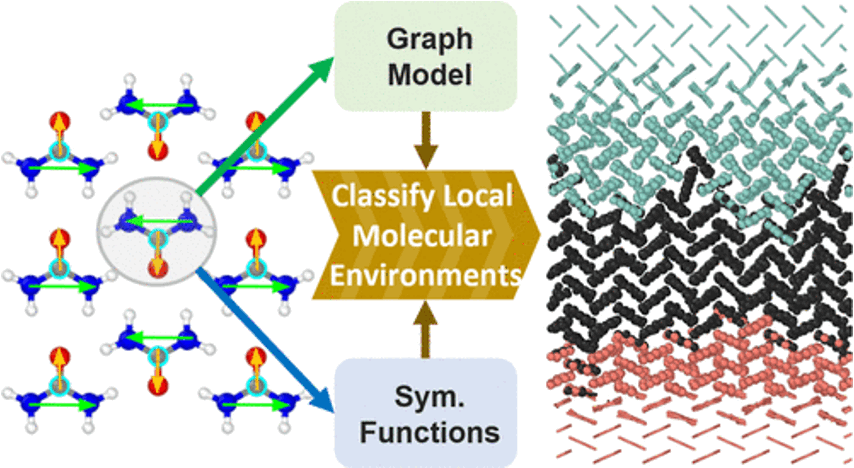
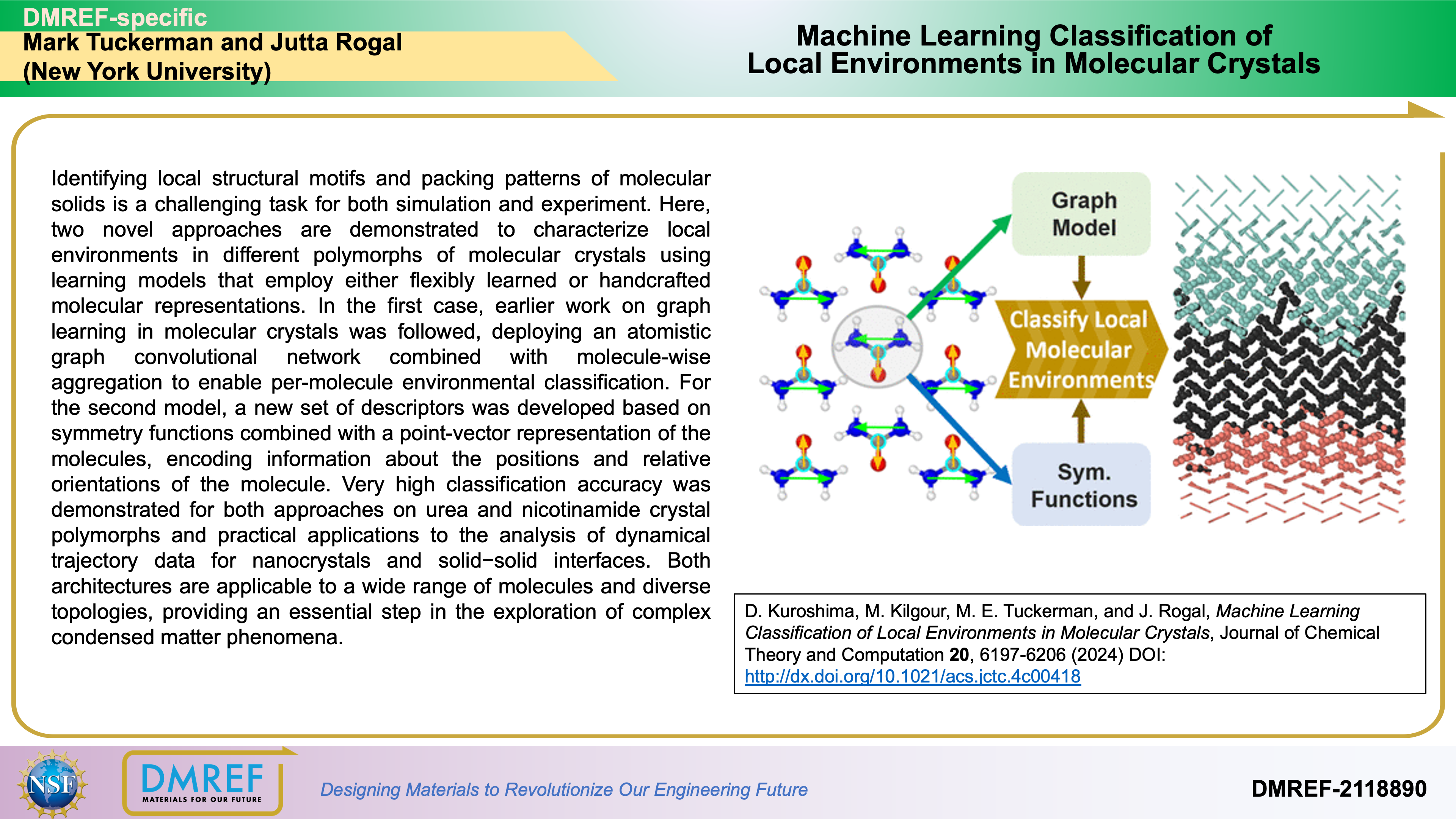
Identifying local structural motifs and packing patterns of molecular solids is a challenging task for both simulation and experiment. Here, two novel approaches are demonstrated to characterize local environments in different polymorphs of molecular crystals using learning models that employ either flexibly learned or handcrafted molecular representations.
In the first case, earlier work on graph learning in molecular crystals was followed, deploying an atomistic graph convolutional network combined with molecule-wise aggregation to enable per-molecule environmental classification. For the second model, a new set of descriptors was developed based on symmetry functions combined with a point-vector representation of the molecules, encoding information about the positions and relative orientations of the molecule.
Very high classification accuracy was demonstrated for both approaches on urea and nicotinamide crystal polymorphs and practical applications to the analysis of dynamical trajectory data for nanocrystals and solid−solid interfaces. Both architectures are applicable to a wide range of molecules and diverse topologies, providing an essential step in the exploration of complex condensed matter phenomena.