Design Rules to Optimize the Intermolecular Long-range Packing of Organic Semiconductor Crystals
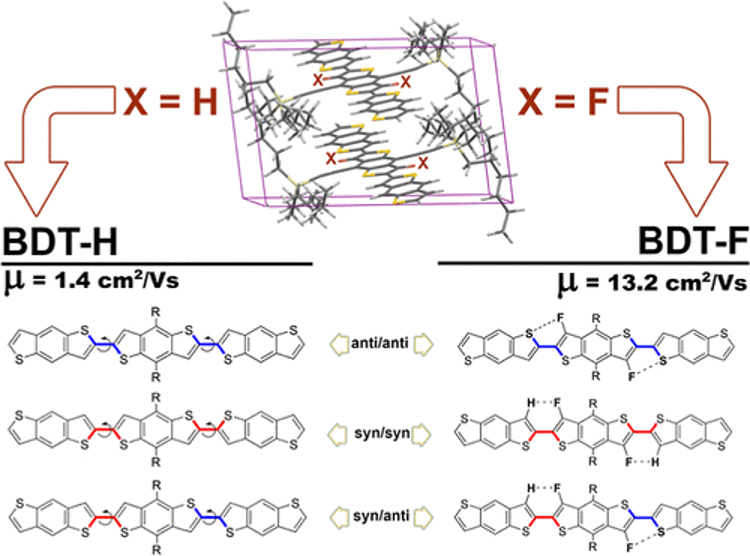
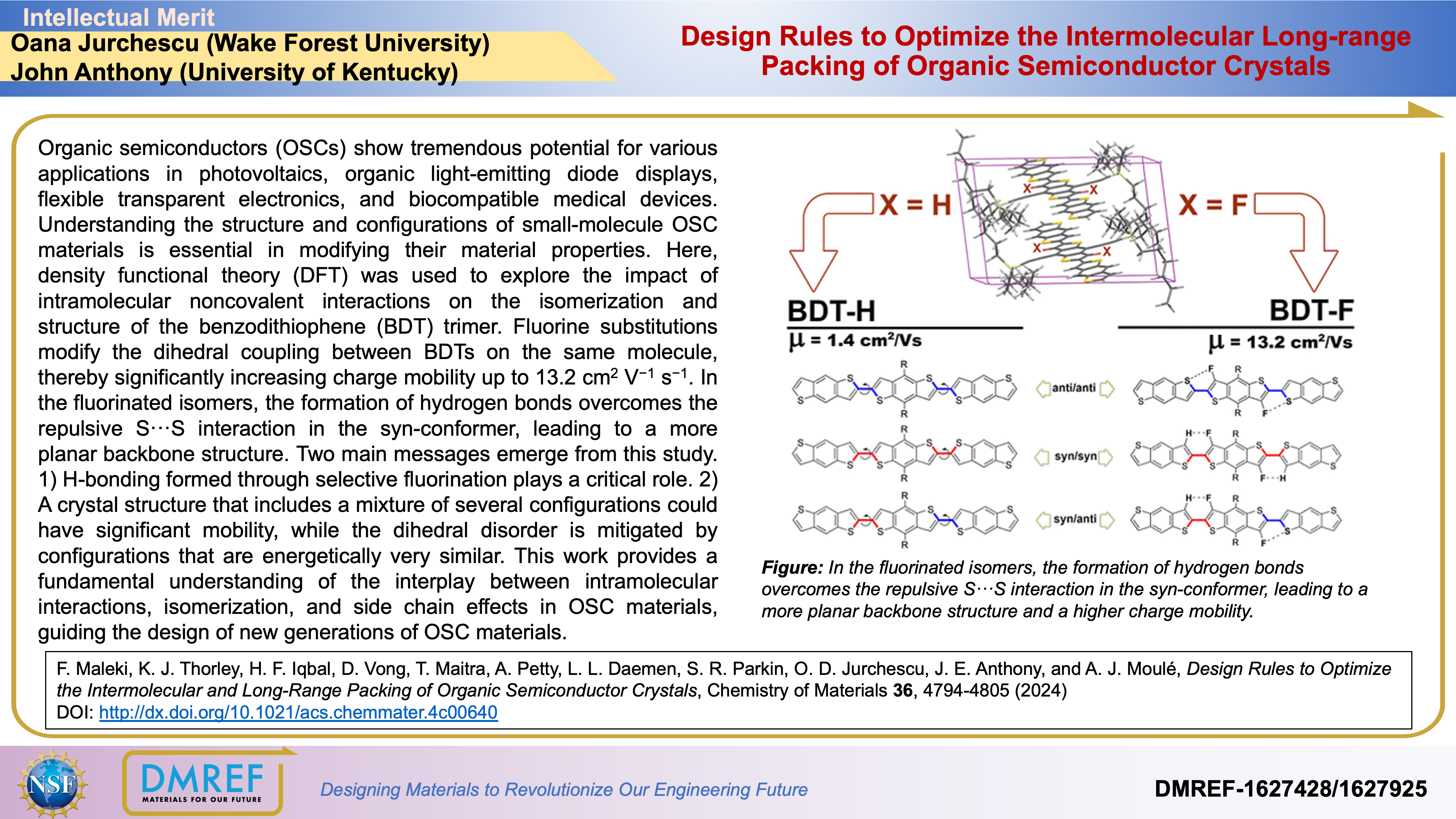
Organic semiconductors (OSCs) show tremendous potential for various applications in photovoltaics, organic light-emitting diode displays, flexible transparent electronics, and biocompatible medical devices. Understanding the structure and configurations of small-molecule OSC materials is essential in modifying their material properties.
Here, density functional theory (DFT) was used to explore the impact of intramolecular noncovalent interactions on the isomerization and structure of the benzodithiophene (BDT) trimer. Fluorine substitutions modify the dihedral coupling between BDTs on the same molecule, thereby significantly increasing charge mobility up to 13.2 cm2 V−1 s−1. In the fluorinated isomers, the formation of hydrogen bonds overcomes the repulsive S···S interaction in the syn-conformer, leading to a more planar backbone structure.
Two main messages emerge from this study:
H-bonding formed through selective fluorination plays a critical role
A crystal structure that includes a mixture of several configurations could have significant mobility, while the dihedral disorder is mitigated by configurations that are energetically very similar.
This work provides a fundamental understanding of the interplay between intramolecular interactions, isomerization, and side chain effects in OSC materials, guiding the design of new generations of OSC materials.